-
Torsional Disorder, Symmetry Breaking, and the Crystal Violet Shoulder Controversy
J. Sissaoui, D.S. Budkina and E Vauthey
Journal of Physical Chemistry Letters, 14 (2023), p5602-5606


DOI:10.1021/acs.jpclett.3c01038 | unige:169497 | Abstract | Article HTML | Article PDF | Supporting Info
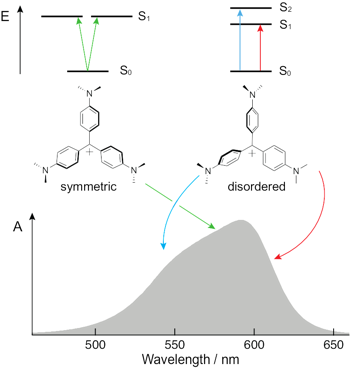
The nature of the lowest-energy electronic absorption band of crystal violet (CV) and particularly the origin of its high-energy shoulder have been debated since the middle of the past century. The most recent studies invoke a splitting of the S1 state upon symmetry breaking induced by interactions with the solvent and/or the counterion. Using a combination of stationary and time-resolved polarized spectroscopy together with quantum-chemical calculations, we show that torsional disorder in the ground-state results in an inhomogeneous broadening of the absorption band of CV. The center of the band is mostly due to symmetric molecules with a degenerate S1 state, whereas the edges originate from transitions to the S1 and S2 states of distorted symmetry-broken molecules. Transient-absorption measurements with different excitation wavelengths reveal that these two groups of molecules interconvert rapidly in liquid but not in a rigid environment.